MRD
1. Introduction to MRD
MRD (Mutation Region Detection) is a software package for finding mutation regions where the input individuals are closely related, but the pedigree is not given. MRD takes the genotype data on a chromosome as well as the disease status for a set of input individuals without any pedigree information and outputs the predicted mutation regions.
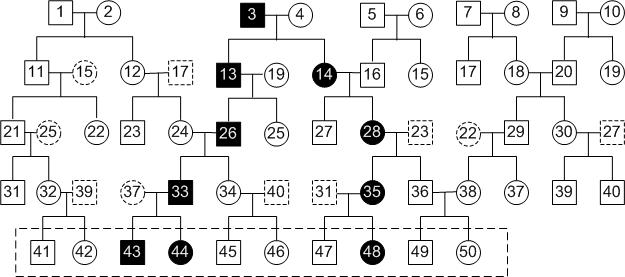
A typical example is shown in the following figure.
In this pedigree, there are 5 generations and 10 founders. We assume that we only know the genotype data of the 10 individuals in the dashed rectangle at the bottom of the pedigree. In this example, the input of our program consists of the genotype data of the 10 individuals in the dashed rectangle at the bottom as well as the disease status (diseased or normal) for the 10 individuals. The pedigree structure and the genotype data for other individuals are not known.
In practice, one can also know the genotype data as well as the disease status for individuals in the latest two or three generations. In this case, those individuals in the latest two or three generations can be treated in the same way as the individuals in the latest generation and put in the set of input individuals.
2. Download
The package is implemented in Java, and we provide a jar package for download which is "MRD.jar". Currently the package is only for Windows. Click MRD to download the package. To run our program MRD, JRE is required to be installed on the machine.
3. How to USE
Double click "simulation.exe", you can run the simulation program that generates simulated input examples for testing MRD.
Go to the directory of the jar package and use the command
>java -Xms32m -Xmx1000m -jar MRD.jar -g genotype.txt -d diseased-status.txt
to run the program.
Here "genotype.txt" is the input genotype file, and "diseased-status.txt" is the input file containing the disease status of the individuals.
Also note that "-Xms32m" and "-Xmx1000m" are parameters for Java Virtual Machine, which allocate memory for the program with minimum 32MB and maximum 1000MB.
4. Input Files
MRD needs the following two basic input files.
(1)genotype.txt
This file contains the genotype data of the individuals in the pedigree
except the founders. The first line includes the tags for each column of the file, which are rsID, snp_position, and the id for each individual. The following lines are the genotype data for each individual on each position. The first two columns are the rsID and the positions of the SNPs, and after that each column stores the genotype data for each individual
at that position. The following is an example of a genotype file.
rsID snp_position 31 32 33
rs1256480 724325 AA AA AA
rs3131972 742584 GA AG AG
rs3131969 744045 GA AG AG
rs3131967 744197 CT TC TC
rs1048488 750775 TT CT CT
When MRD is running, users can specify the id of the input individuals and MRD will select the genotype data for the input individuals from this file. MRD also allows the users to provide their own genotype files. In this case, the file can only contain the genotype data of the input individuals.
(2)diseased-status.txt
This file contains the id and disease status of each individual in the the pedigree except the founders, 0 for diseased and 1 for normal. Users can also provide their own files other than that generated by the simulation program.
5. Simulation Program
We provide a simulation program to generate the genotype data of the input individuals for the package. The program takes a pedigree and two haplotype segments of each founder in the pedigree on the whole chromosome as input. It generates the two copies of haplotype segments for each of the other individuals in the pedigree using the standard chi-square model for recombination with m equals 4 and according to male/female averaged genetic map for chromosome 1 downloaded from HapMap(http://hapmap.org).
The simulation program needs three input files:
(1)hapmap_chr1.phased
This file contains the haplotype data for chromosome 1 of 170 unrelated Japanese in Tokyo and Han Chinese in Beijing, China.
(2)genetic_map_chr1.txt
This file provides the genetic map for the whole chromosome. It contains the physical loci information for the SNP markers in "hapmap_chr1.phased". It can also be downloaded from HapMap(http://hapmap.ncbi.nlm.nih.gov/).
(3)pedigree.ped
This file contains the pedigree. The first row contains the general information, including the number of all individuals, the number of generations and the number of individuals in each generation. For the following rows, each corresponds to an individual in the pedigree. each row contains five columns which stand for the individual's name, father's name(-1 if unavailable ), mother’s name(-1 if unavailable), gender(0 for male, 1 for female), and disease status(0 for diseased individual, 1 for normal individual).
The following is an example file containing three individuals A, B, C.
3 2 2 1
A -1 -1 0 0
B -1 -1 1 1
C A B 0 0
In this file, C is the child of A, B. A and C are diseased while B is normal.
These three files are in the directory of "input for simulation". To run the simulation program, double-click "simulation.exe". The program will randomly select the founders from the 170 individuals in "hapmap_chr1.phased". The simulation program will produce the input files "genotype.txt" and "diseased-status.txt" for MRD as well as the other two files "diseased-haplotype.txt" and "ancestor.txt" for the purpose of computing precision and recall.
6. Computing Precision and Recall
To compute the precision and recall, we should know the real mutation region. We need the following files as part of the input of MRD.
(1)diseased-haplotype.txt: contains the id of the individuals and the status of haplotype for each individual, where 0 indicates that the 0th haplotype is diseased, 1 indicates that the 1th haplotype is diseased and -1 indicates that neither of the two haplotypes is diseased.
(2)ancestor.txt: contains the ancestor information for each haplotype of the individuals except the founders. It is generated by the simulation program. In the simulation program, the founders are used to generate the descendents in the pedigree. Therefore, the haplotype of the founder from which the haployptes for the descendents inherit are known in the process of simulation. This file keeps this information. The first line includes the tags for each column of the file, which are rsID, snp_position, and the id for each individual. The following lines are the ancestor information for each individual on each position. From the third column, each two columns stand for the haplotype of the founders the particular individual inherit from.
7. Run MRD with Simulation Data
To run MRD with simulation data, use the following command:
>java -Xms32m -Xmx1000m -jar MRD.jar -g genotype.txt -d diseased-status.txt -h diseased-haplotype.txt -a ancestor.txt.
The line options are as follows:
[-g]: genotype file
[-d]: disease status file
[-h]: the file contains the disease status for the haplotypes of each individual
[-a]: the ancestor file.
With the "-h" and "-a" options, the precision and recall will be computed by MRD.
All the command options listed above are optional. If no options are used, the program will choose the default files as the input which is equivalent to using the following command:
>java -Xms32m -Xmx1000m -jar MRD.jar -g genotype.txt -d diseased-status.txt .
8. Output
Without using the two options "-h" and "-a", the package will return the results in the file "z-result-1.txt". The results contain the at most 3 regions output by the algorithm. The following is a typical output.
The detected mutation regions are:
first_longest length second_longest length third_longest length
[85641,90864] 5224 [0,0] 0 [0,0] 0
Time Start: Sun Jun 26 21:05:21 CST 2011, Time End: Sun Jun 26 21:05:35 CST 2011
The used time is 14.24 seconds.
Using the two options "-h" and "-a", the precision and recall will be computed. And the output will be in the file "z-result-2.txt". A typical output in this case is as follows:
The real mutation regions are: [85644,90855]
The final results are:
real_region-0 first_longest length second_longest length third_longest length
precision recall
[85644,90855] [85641,90864] 5224 [0,0] 0 [0,0] 0 0.997703 1.000000
Time Start: Sun Jun 26 21:03:23 CST 2011, Time End: Sun Jun 26 21:03:41 CST 2011
The used time is 18.00 seconds.
9. A Real Case Study
We also do experiments using the real data from phase II HapMap database to test our program. The data is about two CEU (Utah residents with European ancestry from the CEPH collection) families (parent-offspring trios) CEPH 1341 and CEPH 1375. You can click here to download the input and output for the real case study in the paper. The input is in the directory "input". You can use
>java -Xms32m -Xmx1000m -jar MRD.jar"
to run the program in the directory of "Real Case". We also output the shared segment of the four diseased individuals which is in the file "sharedSegment.txt".
10. Supplementary Material
You can click here to download the supplementary material of the paper.