Start SEGID from the webpage by clicking the button "start SEGID", a window will pop up:
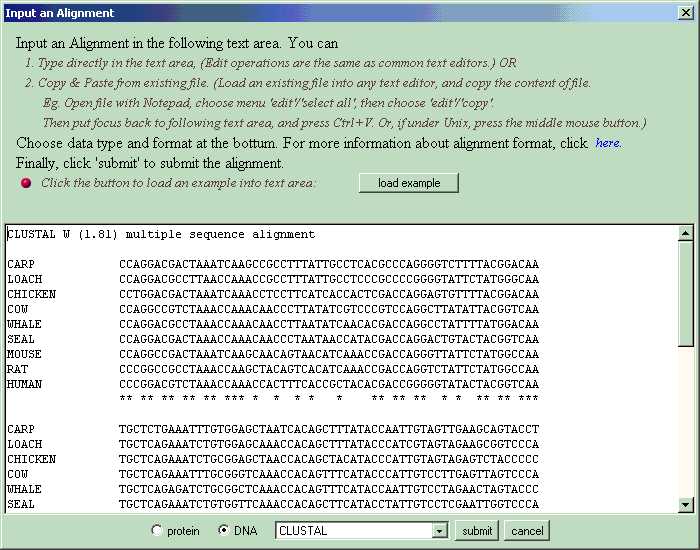
Now, input a multiple
sequence alignment to get it work. Click button 'Input', then click button
'Input Alignment' in the pop-up dialog. Or, choose menu 'input'>'input
Alignment'. Now you can an input dialog. Input the alignment in the text
area. You may
1. Type directly in the textarea, (Edit operations are the same as common
text editors.) OR
2. Copy & Paste from an existing file. (If you have a file containing
the alignment, load the file in any
text editor, and copy the content of file. Eg. open the file with Notepad,
choose menu 'edit'>
'select all', then choose menu 'edit'>'copy' to copy the alignment data
you want to input. Now, return
to SEGID and move focus to the textarea in input dialog by clicking in
the textarea. Press Ctrl+V
to paste alignment. Or, if under Unix, click the middle button of mouse.)
Choose 'protein' or 'DNA' according to your data, also choose the appropriate data format.
Finally, click 'submit' to submit the alignment.
The
alignment format SEGID recognizes includes FASTA, CLUSTAL, GCG-MSF, and
Stockholm. For each format, an example alignment is provided. It can be
loaded into input textarea by clicking button 'load example' in the input
dialog and then choosing corresponding format. For example, following is
a multiple sequence alignment of Clustal format including 9 sequences.
CLUSTAL W (1.81) multiple sequence
alignment
CARP
CCAGGACGACTAAATCAAGCCGCCTTTATTGCCTCACGCCCAGGGGTCTTTTACGGACAA
LOACH CCAGGACGCCTTAACCAAACCGCCTTTATTGCCTCCCGCCCCGGGGTATTCTATGGGCAA
CHICKEN CCTGGACGACTAAATCAAACCTCCTTCATCACCACTCGACCAGGAGTGTTTTACGGACAA
COW
CCAGGCCGTCTAAACCAAACAACCCTTATATCGTCCCGTCCAGGCTTATATTACGGTCAA
WHALE CCAGGACGCCTAAACCAAACAACCTTAATATCAACACGACCAGGCCTATTTTATGGACAA
SEAL
CCAGGACGACTAAACCAAACAACCCTAATAACCATACGACCAGGACTGTACTACGGTCAA
MOUSE CCAGGCCGACTAAATCAAGCAACAGTAACATCAAACCGACCAGGGTTATTCTATGGCCAA
RAT
CCCGGCCGCCTAAACCAAGCTACAGTCACATCAAACCGACCAGGTCTATTCTATGGCCAA
HUMAN CCCGGACGTCTAAACCAAACCACTTTCACCGCTACACGACCGGGGGTATACTACGGTCAA
** ** ** ** ** *** * * * * *
** ** ** * * ** ** ***
CARP
TGCTCTGAAATTTGTGGAGCTAATCACAGCTTTATACCAATTGTAGTTGAAGCAGTACCT
LOACH TGCTCAGAAATCTGTGGAGCAAACCACAGCTTTATACCCATCGTAGTAGAAGCGGTCCCA
CHICKEN TGCTCAGAAATCTGCGGAGCTAACCACAGCTACATACCCATTGTAGTAGAGTCTACCCCC
COW
TGCTCAGAAATTTGCGGGTCAAACCACAGTTTCATACCCATTGTCCTTGAGTTAGTCCCA
WHALE TGCTCAGAGATCTGCGGCTCAAACCACAGTTTCATACCAATTGTCCTAGAACTAGTACCC
SEAL
TGCTCAGAAATCTGTGGTTCAAACCACAGCTTCATACCTATTGTCCTCGAATTGGTCCCA
MOUSE TGCTCTGAAATTTGTGGATCTAACCATAGCTTTATGCCCATTGTCCTAGAAATGGTTCCA
RAT
TGCTCTGAAATTTGCGGCTCAAATCACAGCTTCATACCCATTGTACTAGAAATAGTGCCT
HUMAN TGCTCTGAAATCTGTGGAGCAAACCACAGTTTCATGCCCATCGTCCTAGAATTAATTCCC
***** ** ** ** ** * ** ** ** * ** ** ** ** * **
**
CARP
CTCGAACACTTCGAAAAC---------------------TGATCCTCATTAATACTAGAA
LOACH CTATCTCACTTCGAAAAC---------------------TGGTCCACCCTTATACTAAAA
CHICKEN CTAAAACACTTTGAAGCC---------------------TGATCCTCACTA---------
COW
CTAAAGTACTTTGAAAAA---------------------TGATCTGCGTCAATATTA---
WHALE CTAGAAGTCTTTGAAAAA---------------------TGATCTGTATCAATACTA---
SEAL
CTATCCCACTTCGAGAAA---------------------TGATCTACCTCAATGCTT---
MOUSE CTAAAATATTTCGAAAAC---------------------TGATCTGCTTCAATAATT---
RAT
CTAAAATATTTCGAAAAC---------------------TGATCAGCTTCTATAATT---
HUMAN CTAAAAATCTTTGAAATA---------------------GGGCCCGTATTTACCCTATAG
** ** **
* *
CARP
GACGCCTCGCTAGGAAGCTAA
LOACH GACGCCTCACTAGGAAGCTAA
CHICKEN ---------CTGTCATCTTAA
COW
------------------TAA
WHALE ------------------TAA
SEAL
------------------TAA
MOUSE ------------------TAA
RAT
------------------TAA
HUMAN ---------------------
You can load it by clicking button
'load example' in the input dialog, and then choose 'Clustal' in pop-up
dialog.
SEGID reads the alignment, calculates a score for every column with chosen scoring scheme (default scoring is SP-score and IDENTITY matrix. Users can specify scoring method and matrix via 'set scoring scheme'.) Then the alignment is displayed, and conserved segments (high score substrings) are identified. Three algorithms for identifying conserved segments are provided.
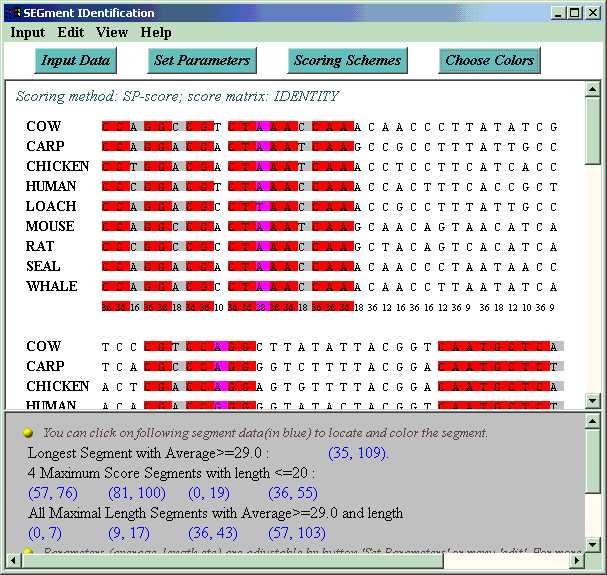
By default, all maximal length segments with average value and length lower bound are colored with pink in the alignment, among which columns of particularly poor scores (below the threshold set by user) are colored with lightgray, and good columns are colored with magenta. Users can also switch to other algorithms of interesting by clicking button 'choose algorithm' or choosing menu 'view'>'algorithm'. Accurate positions of all these segments are output at the bottom of the window. Users can locate a segment in the alignment by clicking on the segment position data.
For more information about how to use this software, please refer to "help" menu or button in SEGID.